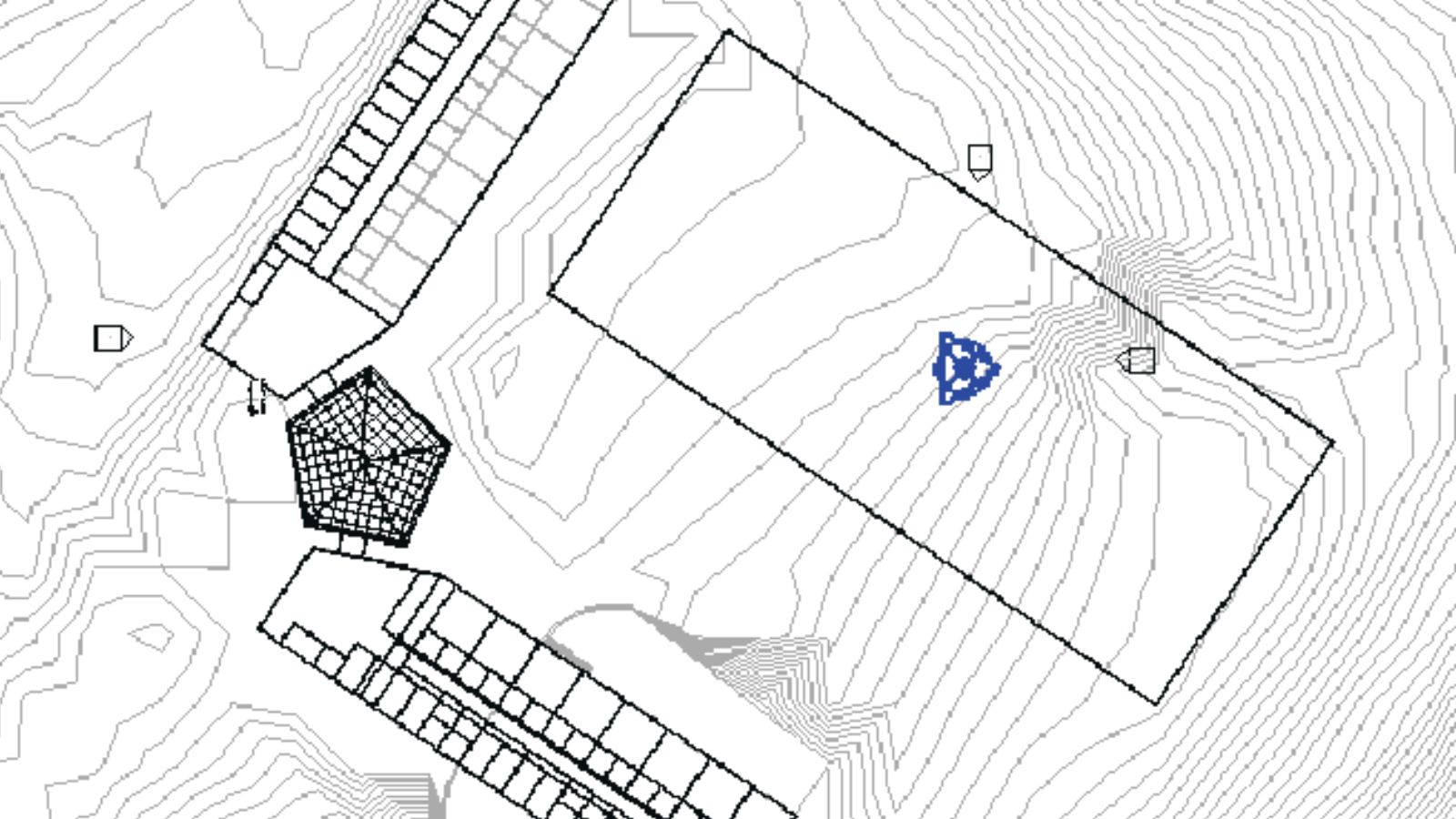
‘Embrace the Ditch,’ and Other Lessons Learned in Duke CEE’s Overture Engineering
Civil and environmental engineering students learn to design buildings within less-than-optimal parameters in a collaborative capstone course
We’re sorry, but that page was not found or has been archived. Please check the spelling of the page address or use the site search.
Still can’t find what you’re looking for? Contact our web team »
Read stories of how we’re teaching students to develop resilience, or check out all our recent news.
Civil and environmental engineering students learn to design buildings within less-than-optimal parameters in a collaborative capstone course

On a Star Wars-themed field of play, student teams deployed small robots they had constructed

Two projects from First-Year Design course are patent-pending. Student surveys suggest the course also fosters teamwork, leadership and communication skills.
May 23
Come on over to the Devil’s Krafthouse at the Brodhead Center for snacks, drinks, and a fun time with your Pratt friends and colleagues celebrating another great year. RSVP by […]
4:00 pm – 4:00 pm
May 24
Medical robots can precisely manipulate tools beyond human capabilities and are thus helpful for surgeries involving delicate tissue interactions. When coupled with live 3D imaging, such robots can independently guide […]
12:00 pm – 1:00 pm Teer 203
May 27