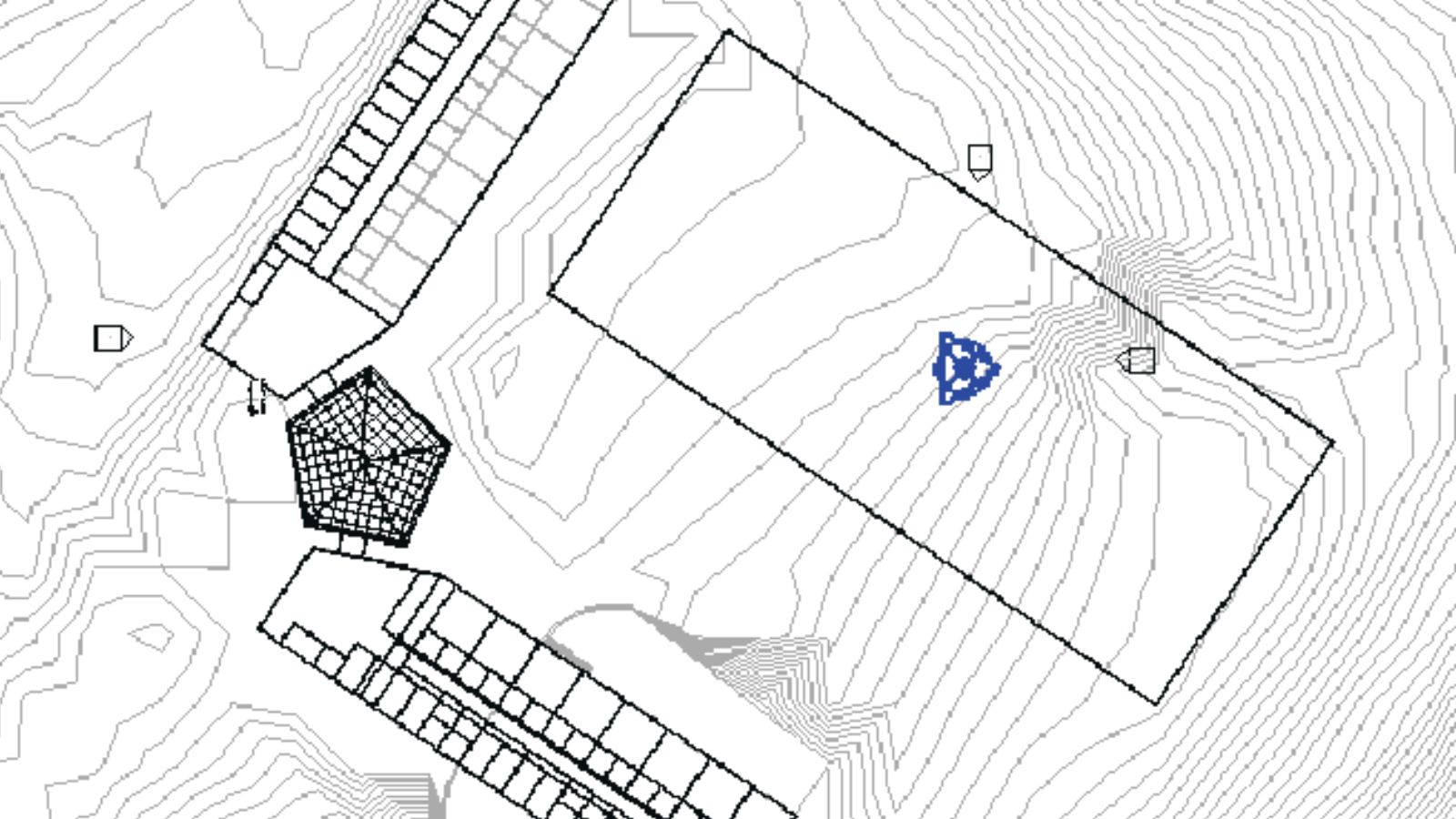
‘Embrace the Ditch,’ and Other Lessons Learned in Duke CEE’s Overture Engineering
Civil and environmental engineering students learn to design buildings within less-than-optimal parameters in a collaborative capstone course
We’re sorry, but that page was not found or has been archived. Please check the spelling of the page address or use the site search.
Still can’t find what you’re looking for? Contact our web team »
Read stories of how we’re teaching students to develop resilience, or check out all our recent news.
Civil and environmental engineering students learn to design buildings within less-than-optimal parameters in a collaborative capstone course

On a Star Wars-themed field of play, student teams deployed small robots they had constructed

Two projects from First-Year Design course are patent-pending. Student surveys suggest the course also fosters teamwork, leadership and communication skills.
Apr 16
The rapid evolution of large language models (LLMs) has ushered in a new era of computational linguistics, yet a systematic approach to their evaluation, particularly in sensitive domains such as […]
12:00 pm – 12:00 pm Virtual
Apr 16
Thomas Lord Department of Mechanical Engineering & Materials Science welcomes Prof. Dr. Harm-Anton Klok, Ecole Polytechnique Fédérale de Lausanne (EPFL), to present a MEMS Seminar: “”Mechanochemistry of Solvent-Swollen Surface-Grafted Polymer […]
12:00 pm – 12:00 pm Teer 203
Apr 16
Computing education postdoc positions are difficult to find; on top of that, postdoc positions are perceived as challenging. In this session, the three current AiiCE postdocs share their experiences with […]
5:00 pm – 5:00 pm Virtual